Thermodynamics out of equilibrium
#PHYS213
Lecture 7: Thermodynamic Processes Reversible Processes
系统状态的变化路径称为热力学过程。我们特别关注以下四种理想化的过程,它们构成了许多热力学循环的基础:
-
等容过程 (Isochoric Process):
- 定义:体积 $V$ 保持不变 ($dV=0$)。
- P-V 图:一条垂直的线段。
- 热力学第一定律 (First Law of Thermodynamics, FLT): $\Delta U = Q - W_{by}$。
- 做功 (Work done by the system): $W_{by} = \int p dV = 0$。因为体积不变,系统不对外做功。
- 热量 (Heat): $Q = \Delta U$。所有传入的热量都用于增加系统的内能。
- 对于理想气体 (Ideal Gas): $\Delta U = C_V \Delta T = \alpha Nk \Delta T$。因此 $Q = C_V \Delta T$。 (这里 $\alpha$ 取决于气体类型,例如单原子气体 $\alpha=3/2$)。利用理想气体状态方程 $pV=NkT$,在体积不变时,$p/T = \text{constant}$,所以 $\Delta p / \Delta T = \text{constant}$,可以得到 $\Delta U = \alpha V \Delta p$ (如 Slide 8 所示)。
-
等压过程 (Isobaric Process):
- 定义:压强 $p$ 保持不变 ($dp=0$)。
- P-V 图:一条水平的线段。
- 做功: $W_{by} = \int p dV = p \int dV = p (V_f - V_i) = p \Delta V$。
- 内能变化 (Internal Energy Change): $\Delta U = C_V \Delta T$ (对于理想气体,内能仅是温度的函数)。
- 热量: $Q = \Delta U + W_{by} = C_V \Delta T + p \Delta V$。
- 对于理想气体: $p \Delta V = Nk \Delta T$。所以 $Q = C_V \Delta T + Nk \Delta T = (C_V + Nk) \Delta T$。我们定义 定压热容 (Heat capacity at constant pressure) $C_p = C_V + Nk$。因此 $Q = C_p \Delta T$。
- 技术比喻: 等压加热时,系统像一个需要推开活塞才能膨胀的气球。一部分热量用来提高内部温度(增加内能 $\Delta U$),另一部分则用来对外做功 $W_{by}$ 把活塞推开。因此,等压过程比等容过程需要更多的热量来达到相同的温度升高,即 $C_p > C_V$ (Slide 5)。
-
等温过程 (Isothermal Process):
- 定义:温度 $T$ 保持不变 ($dT=0$)。
- P-V 图:一条双曲线 ($p \propto 1/V$ 对于理想气体)。
- 对于理想气体: 内能 $U$ 只依赖于 $T$,所以 $\Delta U = 0$。
- 热力学第一定律: $\Delta U = 0 = Q - W_{by}$,因此 $Q = W_{by}$。系统吸收的热量全部用来对外做功。
- 做功 (理想气体): $$ W_{by} = \int_{V_i}^{V_f} p dV = \int_{V_i}^{V_f} \frac{NkT}{V} dV = NkT \int_{V_i}^{V_f} \frac{dV}{V} = NkT \ln\left(\frac{V_f}{V_i}\right) $$
- 这个过程通常需要系统与一个大的 热库 (thermal reservoir) 接触以保持温度恒定。
-
绝热过程 (Adiabatic Process):
- 定义:系统与外界没有热量交换 ($Q=0$)。这通常发生在快速进行的过程中(来不及热交换)或者系统被良好绝热时。
- P-V 图:比等温线更陡峭的曲线。
- 热力学第一定律: $\Delta U = Q - W_{by} = -W_{by}$。系统对外做功会导致内能减少(温度下降),对系统做功会导致内能增加(温度升高)。
- 对于理想气体 (准静态过程 Quasi-static process): 我们可以推导出过程方程 (Slide 6)。从 $\Delta U = -W_{by}$ 开始: $\alpha Nk dT = -p dV = -\frac{NkT}{V} dV$ 分离变量:$\alpha \frac{dT}{T} = -\frac{dV}{V}$ 积分:$\alpha \int \frac{dT}{T} = -\int \frac{dV}{V}$ $\alpha \ln T = -\ln V + \text{constant}$ $\ln(T^\alpha) + \ln V = \text{constant} \Rightarrow \ln(T^\alpha V) = \text{constant}$ 所以: $$ T^\alpha V = \text{constant} $$ 利用 $pV=NkT$ 消去 $T$ ($T \propto pV$),得到: $(pV)^\alpha V = \text{constant} \Rightarrow p^\alpha V^{\alpha+1} = \text{constant}$ 两边取 $1/\alpha$ 次方: $p V^{(\alpha+1)/\alpha} = \text{constant}$。 我们定义 绝热指数 (adiabatic index) $\gamma = C_p/C_V = (C_V+Nk)/C_V = (\alpha Nk + Nk)/(\alpha Nk) = (\alpha+1)/\alpha$。 因此,理想气体准静态绝热过程满足: $$ pV^\gamma = \text{constant} $$ 因为 $\gamma = (\alpha+1)/\alpha > 1$,所以绝热线 $p \propto V^{-\gamma}$ 比等温线 $p \propto V^{-1}$ 更陡峭 (Slide 7)。
可逆性与不可逆性 (Reversibility and Irreversibility)
(Slide 10-14, 26)
-
可逆过程 (Reversible Process): 一个过程是可逆的,如果它可以通过无穷小的改变,使得系统和环境都精确地回到初始状态,并且不留下任何其他变化。
- 关键特征:过程进行得无限缓慢 (准静态,quasi-static),使得系统在每一步都无限接近于平衡态。
- 没有耗散效应 (dissipative effects),如摩擦 (friction)、粘滞性 (viscosity)、非弹性形变、电流产生的热等。
- 没有非平衡的热传递,例如跨越有限温度差的热传递。
- 熵判据 (Entropy Criterion): 一个过程是可逆的,当且仅当系统与环境的总熵变 (total entropy change) 为零:$\Delta S_{tot} = \Delta S_{sys} + \Delta S_{surr} = 0$ (Slide 10)。
- 准静态的绝热过程 ($Q=0$) 是可逆的,因为 $dS = dQ_{rev}/T = 0$,系统熵不变,环境也无变化,$\Delta S_{tot}=0$ (Slide 12)。
- 准静态的等温过程 (与温度恒为 $T$ 的热库接触) 是可逆的。如果系统从热库吸收热量 $Q$,则 $\Delta S_{sys} = Q/T$,热库失去热量 $Q$,$\Delta S_{res} = -Q/T$。因此 $\Delta S_{tot} = Q/T - Q/T = 0$ (Slide 12)。
- Slide 3 和 13 中将等容和等压过程标记为不可逆,这通常是指实际操作中,例如将系统突然与一个不同温度的热库接触进行等容加热,或在有摩擦的活塞下进行等压膨胀。这些实际过程涉及有限温差传热或摩擦,导致 $\Delta S_{tot} > 0$。然而,理想化的等容和等压过程 可以 作为可逆循环的一部分,只要热量交换是通过一系列无穷小的温差或可逆方式进行的。
-
不可逆过程 (Irreversible Process): 自然界中发生的所有宏观过程都是不可逆的。它们不能自行反向进行。
- 特征:过程进行速度有限,系统偏离平衡态;存在耗散效应;存在有限温差传热。
- 总熵总是增加:$\Delta S_{tot} > 0$。
-
例子 (Slide 11, 14, 26):
- 一个物体由于摩擦而减速停止,其宏观动能转化为内能(热)。反过来,物体的内能自发地转化为宏观动能,让物体重新运动起来,是永远不会发生的 (Slide 11)。
- 自由膨胀 (Free Expansion): 气体向真空膨胀 (Slide 14)。这是一个快速、非准静态的过程。虽然对于理想气体 $W=0, Q=0, \Delta U=0$,但过程是高度不可逆的,气体不会自发地收缩回原来的体积。熵是增加的。
- 跨越有限温差的热传递 (热量从热物体传到冷物体)。
- 摩擦生热。
热机与效率 (Heat Engines and Efficiency)
(Slide 15, 22)
- 热机 (Heat Engine): 一种将热能转化为有用功的装置。它通常在一个 循环 (cycle) 中工作,意味着工作物质 (working substance, e.g., gas) 经过一系列过程后回到其初始状态 ($\Delta U_{cycle}=0$)。
-
工作原理 (Slide 15):
- 从高温热库 ($T_h$) 吸收热量 $Q_h$。
- 对外做功 $W_{by}$。
- 向低温热库 ($T_c$) 排放废热 $Q_c$。
- 循环往复。 注意: 热库是指足够大,以至于吸收或放出热量时其自身温度保持不变的系统。
- 能量守恒 (Energy Conservation): 对于一个完整的循环 $\Delta U_{cycle}=0$,根据热力学第一定律 $Q_{net} = W_{by}$。净吸收的热量 $Q_{net} = Q_h - Q_c$ (我们定义 $Q_h, Q_c, W_{by}$ 均为正值,代表其大小)。所以: $$ W_{by} = Q_h - Q_c $$
- 效率 (Efficiency) $\epsilon$ (Slide 22): 定义为 “得到的有用功” 与 “付出的代价 (吸收的热量)” 之比。 $$ \epsilon = \frac{W_{by}}{Q_h} = \frac{Q_h - Q_c}{Q_h} = 1 - \frac{Q_c}{Q_h} $$ 效率表示有多少从高温热库吸收的热量被转化为了功。因为必须有废热 $Q_c$ 排出 (后面会看到这是第二定律的要求),所以效率 $\epsilon$ 总是小于 1 (或 100%)。
斯特林循环 (Stirling Cycle) - 热机实例
(Slide 16-21)
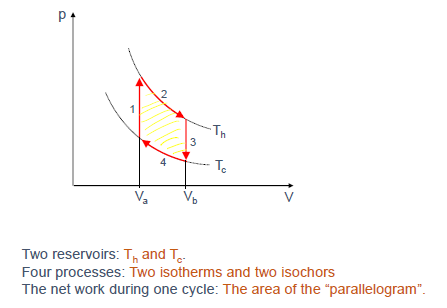
斯特林循环是一个由两个等温过程和两个等容过程组成的理想循环。
- 过程 1 ($4 \to 1$): 等容加热 ($V_a$ 不变, $T$ 从 $T_c$ 升到 $T_h$)。吸收热量 $Q_1 = C_V(T_h-T_c)$。
- 过程 2 ($1 \to 2$): 等温膨胀 ($T_h$ 不变, $V$ 从 $V_a$ 增到 $V_b$)。吸收热量 $Q_2 = W_2 = NkT_h \ln(V_b/V_a)$。
-
过程 3 ($2 \to 3$): 等容冷却 ($V_b$ 不变, $T$ 从 $T_h$ 降到 $T_c$)。放出热量 $ Q_3 = C_V(T_h-T_c)$。 -
过程 4 ($3 \to 4$): 等温压缩 ($T_c$ 不变, $V$ 从 $V_b$ 减到 $V_a$)。放出热量 $ Q_4 = W_4 = NkT_c \ln(V_b/V_a)$。 - 净功 (Net Work): $W_{by} = W_2 + W_4 = NkT_h \ln(V_b/V_a) - NkT_c \ln(V_b/V_a) = Nk(T_h - T_c) \ln(V_b/V_a)$ (Slide 19)。这等于 P-V 图上封闭曲线所围的面积。
- 总吸热 $Q_h$: 来自高温过程的热量。在 Slide 20 中定义为 $Q_h = Q_1 + Q_2 = \alpha Nk(T_h-T_c) + NkT_h \ln(V_b/V_a)$。 (在实际的带回热器 (regenerator) 的斯特林发动机中,$Q_1$ 的热量可以由 $Q_3$ 放出的热量提供,此时 $Q_h$ 仅为 $Q_2$。但按此幻灯片的计算方式,我们用 $Q_1+Q_2$)。
- 效率 (Efficiency): $\epsilon = W_{by}/Q_h = \frac{Nk(T_h - T_c) \ln(V_b/V_a)}{\alpha Nk(T_h-T_c) + NkT_h \ln(V_b/V_a)}$。
- 数值算例 (Slide 21): 对于 $V_b=2V_a$, 单原子气体 $\alpha=3/2$, $T_h=373K$, $T_c=273K$,计算得到 $\epsilon \approx 16.9\%$。
热力学第二定律与卡诺效率 (The Second Law and Carnot Efficiency)
(Slide 23-27, 31)
-
热力学第二定律 (The Second Law of Thermodynamics): 有多种等价表述。这里用到的是克劳修斯表述的推论和熵表述:
- 开尔文-普朗克表述推论: 不可能制造出一种循环工作的热机,只从一个热源吸热,将之完全转化为功,而不在低温热库留下任何其它变化 (即 $Q_c$ 不可能为零)。
- 熵表述: 在任何涉及孤立系统的过程中,系统的总熵永不减少 ($\Delta S_{tot} \ge 0$)。对于可逆过程 $\Delta S_{tot}=0$,对于不可逆过程 $\Delta S_{tot}>0$。
-
效率上限 (Maximum Efficiency): (Slide 23-24)
考虑任意一个在 $T_h$ 和 $T_c$ 之间工作的热机。在一个循环中,引擎本身熵变 $\Delta S_{engine}=0$。热库的熵变是 $\Delta S_{hot} = -Q_h/T_h$ (失去热量) 和 $\Delta S_{cold} = +Q_c/T_c$ (得到热量)。
根据第二定律,总熵变 $\Delta S_{tot} = \Delta S_{engine} + \Delta S_{hot} + \Delta S_{cold} = 0 - Q_h/T_h + Q_c/T_c \ge 0$。
整理得到:$\frac{Q_c}{T_c} \ge \frac{Q_h}{T_h}$,即 $\frac{Q_c}{Q_h} \ge \frac{T_c}{T_h}$。
将此代入效率公式 $\epsilon = 1 - Q_c/Q_h$,得到:
$$
\epsilon \le 1 - \frac{T_c}{T_h}
$$
这个结果表明,任何在给定温度 $T_h$ 和 $T_c$ 之间工作的热机,其效率不可能超过 $1 - T_c/T_h$。这个上限被称为 卡诺效率 (Carnot Efficiency):
$$
\epsilon_{Carnot} = 1 - \frac{T_c}{T_h}
$$
- 这是一个 普适定律 (universal law),不依赖于工作物质或热机的具体设计。
- 只有 可逆热机 (reversible engine) 才能达到卡诺效率 (此时 $\Delta S_{tot}=0$,不等号取等号)。
- 提高效率的方法:提高 $T_h$ 或降低 $T_c$ (Slide 25)。
-
卡诺循环 (Carnot Cycle): (Slide 27)
为了达到卡诺效率,热机必须只由可逆过程组成。卡诺设计了一个理想的可逆循环,由以下四个过程组成:
- 等温膨胀 (Isothermal expansion) at $T_h$。
- 绝热膨胀 (Adiabatic expansion) (温度从 $T_h$ 降到 $T_c$)。
- 等温压缩 (Isothermal compression) at $T_c$。
- 绝热压缩 (Adiabatic compression) (温度从 $T_c$ 回到 $T_h$)。 由于所有过程都是可逆的,卡诺循环的效率等于 $\epsilon_{Carnot}$。
- 效率损失与熵产生 (Efficiency Loss and Entropy Production): (Slide 26, 31) 对于任何不可逆的热机 ($\Delta S_{tot} > 0$),其效率必然低于卡诺效率。效率损失的大小与总熵产生 $\Delta S_{tot}$ 直接相关: $$ \epsilon = \epsilon_{Carnot} - \frac{T_c \Delta S_{tot}}{Q_h} $$ 要提高效率,就要尽量减少系统中的不可逆性(减少熵产生)。
制冷机与热泵 (Refrigerators and Heat Pumps)
(Slide 28-30)
热机是利用温差做功,反过来,我们可以利用功来传递热量,这就是制冷机和热泵。它们的工作原理本质上是热机循环的逆转。
-
性能系数 (Coefficient of Performance, COP): 用于衡量制冷机和热泵的性能,定义为 “得到的结果” / “付出的代价 (输入的功)”。
-
制冷机 (Refrigerator):
- 目标:从低温处 ($T_c$) 移走热量 $Q_c$ (使其保持低温)。
- 代价:需要输入功 $W_{in}$ (注意 $W_{in}$ 在图中标记为 $W_{on}$)。
- 能量守恒:$Q_h = Q_c + W_{in}$ (向高温环境 $T_h$ 排出总热量 $Q_h$)。
- 性能系数:$\text{COP}{Ref} = K = \frac{Q_c}{W{in}}$。
- 卡诺制冷机制冷系数 (上限): $$ K_{Carnot} = \frac{T_c}{T_h - T_c} $$
-
热泵 (Heat Pump):
- 目标:向高温处 ($T_h$) 输送热量 $Q_h$ (例如给房间供暖)。
- 代价:需要输入功 $W_{in}$。
- 能量守恒:$Q_h = Q_c + W_{in}$ (从低温环境 $T_c$ 吸收热量 $Q_c$)。
- 性能系数:$\text{COP}{HP} = K’ = \frac{Q_h}{W{in}}$。
- 卡诺热泵制热系数 (上限): $$ K’_{Carnot} = \frac{T_h}{T_h - T_c} $$
- 关系:$\text{COP}{HP} = \text{COP}{Ref} + 1$。
- 重要特点: 当温差 $T_h - T_c$ 很小时,制冷机和热泵的 COP 可以远大于 1。这意味着它们传递的热量可以远大于驱动它们所需的功。例如,Slide 30 的例子中,只需 8.5W 的功率就能将 70W 的热量从冰箱内部抽出。
Lecture 8: Reversible Processes
热力学基本概念回顾
- 系统 (System) 与环境 (Environment): 在热力学中,我们通常关注一个特定的系统 (例如,气缸中的气体) 以及它周围的环境 (Surroundings)。
-
热机示意图 (Heat Engine Diagram): Slide 2 展示了一个典型的热机模型。
- 它从高温热源 (Hot Reservoir) $T_H$ 吸收热量 $Q_H$。
- 一部分能量转化为功 (Work) $W_{out}$ 输出。
- 剩余的热量 $Q_C$ 排放到低温热源 (Cold Reservoir) $T_C$。
-
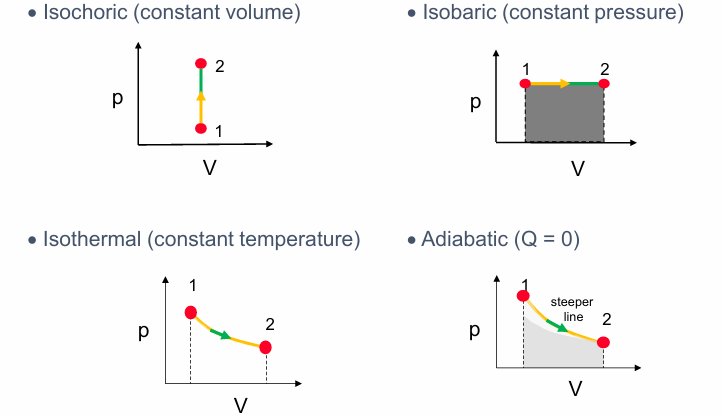
四种基本热力学过程 (Four Thermodynamic Processes, Slide 3):
- 等容过程 (Isochoric Process): 体积 $V$ 保持不变。在 P-V 图上是一条垂直线。
- 等压过程 (Isobaric Process): 压强 $p$ 保持不变。在 P-V 图上是一条水平线。过程中做的功是 $W = p \Delta V$,在 P-V 图上是曲线下的面积 (灰色区域)。
- 等温过程 (Isothermal Process): 温度 $T$ 保持不变。对于理想气体,P-V 图上是一条双曲线 ($p \propto 1/V$)。
- 绝热过程 (Adiabatic Process): 系统与外界没有热量交换 ($Q=0$)。对于理想气体,P-V 图上也是一条曲线 ($p \propto 1/V^\gamma$),通常比等温线更陡峭 (steeper)。
可逆过程与熵 (Reversible Processes and Entropy)
- 可逆过程的定义: 一个过程是可逆的,意味着系统和环境总能恢复到初始状态,并且在外界不留下任何变化。
-
可逆过程的熵条件 (Entropy Condition for Reversibility): 一个过程是可逆的,其充要条件是系统与环境的总熵变 (Total Entropy Change) 为零。
$$
\Delta S_{TOT} = \Delta S_{system} + \Delta S_{environment} = 0
$$
- 这意味着在可逆过程中,没有净熵产生 (No net entropy is produced!) (Slide 5)。
-
可逆过程的类型 (Types of Reversible Processes, Slide 5): 在热力学中,只有两种基本类型的过程可以是可逆的:
- 准静态绝热过程 (Quasi-static adiabatic processes, $Q=0$)
-
准静态等温过程 (Quasi-static isothermal processes, T constant)
- 准静态 (Quasi-static): 指过程进行得极其缓慢,系统在任何时刻都无限接近于平衡态 (Equilibrium)。这是实现可逆性的必要条件,但不是充分条件 (还需要无摩擦等)。
[!tip] 准静态 vs. 可逆过程 (Quasi-static vs. Reversible Processes)
- 关系:
- 所有可逆过程都必须是准静态的 (All reversible processes are quasi-static) 。因为如果过程不是缓慢进行的,系统内部就会出现不平衡 (如压强差、温度差),导致熵产生,从而不可逆。
- 但并非所有准静态过程都是可逆的 (Not all quasi-static processes are reversible)。例如,一个缓慢进行的有摩擦的过程,虽然是准静态的,但摩擦会产生熵,因此是不可逆的。
- 技术比喻: 缓慢地 (准静态地) 移动一个物体,如果在有摩擦的表面上移动,能量会因摩擦转化为热量 (熵增加),这个过程无法完美倒过来让热量变回机械能并将物体推回原处,所以是不可逆的。只有在理想的无摩擦表面上缓慢移动,才可能是可逆的。
非平衡过程 (Non-equilibrium Processes, Slide 6)
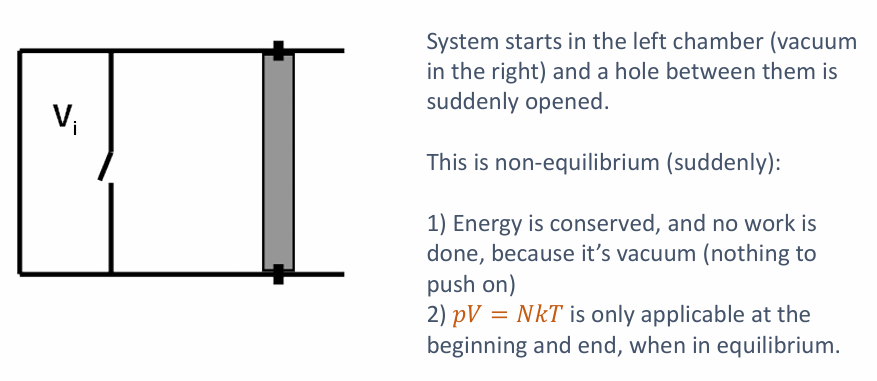
-
自由膨胀 (Free Expansion): Slide 6 的例子是一个典型的非平衡过程。气体突然冲入真空区域。
- 这是一个快速过程,不是准静态的。
- 系统在膨胀过程中并不处于平衡态,因此我们不能用 $pV=NkT$ 这样的状态方程来描述中间过程 (只能描述初始和最终的平衡态)。
- 由于是向真空膨胀,气体不对外做功 ($W=0$)。如果容器绝热 ($Q=0$),根据热力学第一定律 $\Delta U = Q - W = 0$,理想气体的内能不变,因此温度也不变。
- 尽管能量守恒且温度可能不变,但这是一个高度不可逆的过程,总熵是显著增加的 ($\Delta S_{TOT} > 0$)。
绝热与等温过程详解 (Detailed Look at Adiabatic and Isothermal Processes)
-
绝热过程 (Adiabatic Process):
- $Q=0$ (系统与外界无热量交换)。
- 对于可逆 (准静态) 绝热过程,熵变 $\Delta S = \int dQ/T = 0$。系统熵不变。
- 对于理想气体,该过程遵循 $p V^\gamma = \text{const}$,其中 $\gamma = C_p/C_v$ 是绝热指数 (adiabatic index),也等于 $(\alpha+1)/\alpha$,这里 $\alpha = N_{DOF}/2$ (自由度的一半)。
-
等温过程 (Isothermal Process):
- $T = \text{const}$ (系统与一个恒温热库接触)。
- 对于可逆 (准静态) 等温过程,系统与热库之间有热量交换 $Q$。
- 系统熵变 $\Delta S_{sys} = Q/T$
- 热库 (环境) 熵变 $\Delta S_{res} = -Q/T$ (与系统熵变大小相等,符号相反)。
- 总熵变 $\Delta S_{tot} = \Delta S_{sys} + \Delta S_{res} = 0$。
- 对于理想气体,该过程遵循 $p V = \text{const}$ (即 $p = \text{const} / V$)。
热机效率与制冷机制冷系数 (Efficiency and COP)
- 通用评价指标 (General Form): 性能 = 你得到的 (What you get) / 你付出的 (What you paid for)。
-
热机效率 (Engine Efficiency, $\epsilon$):
- 得到: 做出的功 $W$。
- 付出: 从高温热源吸收的热量 $Q_H$ (例如,燃料燃烧产生的热量)。
- $\epsilon = \frac{W}{Q_H}$。根据能量守恒 $W = Q_H - Q_C$,所以 $\epsilon = \frac{Q_H - Q_C}{Q_H} = 1 - \frac{Q_C}{Q_H}$。
-
制冷机性能系数 (Refrigerator COP):
- 得到: 从低温物体 (冰箱内部) 移走的热量 $Q_C$。
- 付出: 消耗的功 $W$ (例如,压缩机消耗的电能)。
- $COP_{ref} = \frac{Q_C}{W}$。
-
热泵性能系数 (Heat Pump COP):
- 得到: 向高温物体 (室内) 输送的热量 $Q_H$。
- 付出: 消耗的功 $W$ (例如,电能)。
- $COP_{HP} = \frac{Q_H}{W}$。
- 注意 $Q_H = W + Q_C$,所以 $COP_{HP} = \frac{W+Q_C}{W} = 1 + COP_{ref}$。热泵的 COP 通常大于 1。
卡诺循环与卡诺效率 (Carnot Cycle and Efficiency, Slide 10, 14, 18)
-
卡诺循环 (Carnot Cycle): 由两个可逆等温过程和两个可逆绝热过程组成的理想热力学循环。
-
卡诺定理 (Carnot’s Theorem): 在相同的高温热源 $T_H$ 和低温热源 $T_C$ 之间工作的所有热机中,可逆热机 (如卡诺热机) 的效率最高。所有可逆热机在该条件下效率都相同。
-
卡诺效率 (Carnot Efficiency, $\epsilon_{Carnot}$):
$$
\epsilon_{Carnot} = 1 - \frac{T_C}{T_H}
$$
- 这是理论上的最大效率。注意:温度 $T_H$ 和 $T_C$ 必须使用绝对温标 (开尔文 Kelvin, K)。
- 对于可逆循环 (卡诺循环),有 $$\frac{Q_C}{Q_H} = \frac{T_C}{T_H}$$
-
不可逆性对效率的影响 (Inefficiency due to Irreversibility):
- 对于任何在 $T_H$ 和 $T_C$ 之间工作的热机,总熵变 $\Delta S_{total} = \frac{Q_C}{T_C} - \frac{Q_H}{T_H} \ge 0$ (等号仅对可逆过程成立)。
- 实际效率 $\epsilon$ 与卡诺效率的关系为: $$ \epsilon = 1 - \frac{Q_C}{Q_H} = 1 - \frac{T_C}{T_H} \left( \frac{Q_C/T_C}{Q_H/T_H} \right) $$ 推导: 从 $\Delta S_{total} = \frac{Q_C}{T_C} - \frac{Q_H}{T_H}$ 可得 $\frac{Q_C}{T_C} = \frac{Q_H}{T_H} + \Delta S_{total}$。 所以 $\frac{Q_C}{Q_H} = \frac{T_C}{T_H} + \frac{T_C \Delta S_{total}}{Q_H}$。 代入效率公式 $\epsilon = 1 - \frac{Q_C}{Q_H}$: $$ \epsilon = 1 - \left( \frac{T_C}{T_H} + \frac{T_C \Delta S_{total}}{Q_H} \right) = \left( 1 - \frac{T_C}{T_H} \right) - \frac{T_C \Delta S_{total}}{Q_H} $$ 即: $$ \epsilon = \epsilon_{Carnot} - \frac{T_C \Delta S_{total}}{Q_H} $$
- 由于 $\Delta S_{total} \ge 0$, $T_C > 0$, $Q_H > 0$,所以实际效率 $\epsilon \le \epsilon_{Carnot}$。任何不可逆因素 ($\Delta S_{total} > 0$) 都会降低热机效率。
- 卡诺热机是理想化的,现实中无法完全实现,因为它要求过程无限缓慢且没有摩擦等损耗。
可选内容: 理想气体绝热过程公式推导 (Optional: Adiabatic formula)
- Slide 20 提供了一个推导理想气体准静态绝热过程 $PV^\gamma = \text{const}$ 的过程。
- 它结合了以下三个基本定律:
- 理想气体状态方程 (Ideal gas law): $pV = NkT$ 及其微分形式 $pdV + Vdp = NkdT$。
- 热力学第一定律 (1st law of Thermo): 对于绝热过程 $dQ=0$,所以 $dU = -pdV$。
- 能量均分定理 (Equipartition theorem): 理想气体内能 $U = \frac{N_{DOF}}{2} NkT$,其微分形式 $dU = \frac{N_{DOF}}{2} NkdT$ (其中 $N_{DOF}$ 是每个分子的自由度)。
- 通过联立和积分这些方程,最终可以得到 $PV^\gamma = \text{const}$,其中 $\gamma = (N_{DOF}/2 + 1) / (N_{DOF}/2) = C_p/C_v$。
好的同学,我们来一起回顾一下 Lecture 9 关于亥姆霍兹自由能 (Helmholtz Free Energy) 的内容。
这次课我们主要讨论了一个新的热力学势——亥姆霍兹自由能,它是联系宏观热力学和微观统计力学的重要桥梁,并且在描述恒温恒容过程系统所能做的最大功方面非常有用。
我会按照讲义的顺序,结合一些例子和解释,来帮你梳理知识点。
Lecture 9: 亥姆霍兹自由能 (Helmholtz Free Energy)
热力学循环回顾 (Review of Thermodynamic Cycles)
1. 热机 (Heat Engine)
(Slides 3, 4)
- 热机是一种将热能转化为功的装置。它从高温热源 ($T_h$) 吸收热量 $Q_h$,一部分转化为功 $W$,另一部分热量 $Q_c$ 排放到低温热源 ($T_c$)。
- 效率 (Efficiency, $\epsilon$): 对所有热机: $$ \epsilon = \frac{W}{Q_h} = 1 - \frac{Q_c}{Q_h} $$
-
卡诺循环 (Carnot Cycle):
- 是一种理想的可逆循环 (Reversible Cycle),由两个等温过程 (Isothermal Process) 和两个绝热过程 (Adiabatic Process) 组成。
- 卡诺热机的效率是所有在相同两个热源之间工作的热机中最高的: $$ \epsilon_{\text{Carnot}} = 1 - \frac{T_c}{T_h} $$
- 对于卡诺循环 (可逆过程): $$ \frac{Q_c}{Q_h} = \frac{T_c}{T_h} $$
-
不可逆性与熵 (Irreversibility and Entropy):
- 实际的热机都是不可逆的 (Irreversible),这意味着在循环过程中总熵会增加 ($\Delta S_{tot} > 0$)。
- 当存在净的熵增时,热机效率会降低: $$ \epsilon = \epsilon_{\text{Carnot}} - \frac{T_c \Delta S_{tot}}{Q_H} $$
2. 绝热/等温过程的可逆性 (Adiabatic/Isothermal processes Reversibility)
-
绝热过程 (Adiabatic Process): 系统与外界没有热量交换 ($dQ=0$)。
- 准静态绝热过程 (Quasi-static adiabatic process) 是可逆的,因为 $\Delta S = \int \frac{dQ}{T} = 0$。
-
等温过程 (Isothermal Process): 系统与恒温热源接触,温度保持不变。
- 对于准静态等温过程,系统从热源吸收 (或放出) 热量 $dQ$。系统熵变 $\Delta S_{sys} = dQ/T$,热源熵变 $\Delta S_{res} = -dQ/T$。
- 总熵变 $\Delta S_{tot} = \Delta S_{sys} + \Delta S_{res} = 0$。因此,准静态等温过程也是可逆的。
3. 热机的逆行:制冷机与热泵 (Heat Engine in Reverse: Refrigerators and Heat Pumps)
- 如果将卡诺循环反向进行,就得到了制冷机或热泵。它们通过外界做功 $W$,将热量从低温热源 ($T_c$) 转移到高温热源 ($T_h$)。
-
性能系数 (Coefficient of Performance, K) (注意这里不用“效率”):
- 制冷机 (Refrigerator): 目的是使低温物体保持低温 (我们关心 $Q_c$)。 $$ K_{\text{refrigerator}} = \frac{Q_c}{W} = \frac{Q_c}{Q_h - Q_c} = \frac{T_c}{T_h - T_c} \quad (\text{对于理想卡诺制冷机}) $$
- 热泵 (Heat Pump): 目的是使高温物体保持高温 (我们关心 $Q_h$)。 $$ K_{\text{heat pump}} = \frac{Q_h}{W} = \frac{Q_h}{Q_h - Q_c} = \frac{T_h}{T_h - T_c} \quad (\text{对于理想卡诺热泵}) $$
- 这里的 $W$ 是我们为制冷机或热泵的运行所付出的代价,比如电费。
4. 制冷的极限 (The Limits of Cooling)
- 从制冷机的性能系数公式 $K_{\text{refrigerator}} = T_c / (T_h - T_c)$ 可以看出,当 $T_c \to 0$ 时,$K \to 0$。
- 这意味着要达到更低的温度,制冷机的性能会急剧下降,需要消耗的功 $W = Q_c/K$ 会急剧增加。
- 结论: 不可能将一个系统冷却到绝对零度 (Absolute Zero)。这是热力学第三定律的一个表述。
可用功与亥姆霍兹自由能 (Available Work and Free Energy)
1. 可用功的推导 (Derivation of Available Work)
- 考虑一个物体 (比如一块砖头,brick) 从初始温度 $T_{brick}$ 冷却到环境温度 $T_{env}$ 。我们想知道一个理想热机能从这个冷却过程中提取多少功 $W_{by}$。
- 根据能量守恒 (热力学第一定律) 和熵的定义 (热力学第二定律,假设卡诺效率),我们得到:
$$
W_{by} = -\Delta U_{brick} + T_{env} \Delta S_{brick}
$$
这里:
- $\Delta U_{brick}$ 是砖头内能 (Internal Energy, $U$) 的变化。砖头降温,$\Delta U_{brick}$ 为负,所以 $-\Delta U_{brick}$ 为正,表示能量被提取出来。
- $\Delta S_{brick}$ 是砖头熵 (Entropy, $S$) 的变化。砖头降温,熵减少,$\Delta S_{brick}$ 为负。$T_{env}\Delta S_{brick}$ 这一项代表了由于熵变,必须传递给环境的热量的一部分,它减少了可对外做的功。
2. 亥姆霍兹自由能的定义 (Definition of Helmholtz Free Energy)
- 为了方便表示上述可用功,我们定义一个新的态函数,称为亥姆霍兹自由能 (Helmholtz Free Energy, $F$): $$ F = U - TS $$ 对于我们讨论的砖块系统,在与温度为 $T_{env}$ 的环境作用时,其自由能可以写为: $$ F_{brick} = U_{brick} - T_{env}S_{brick} $$
- 那么,当系统经历一个过程,其亥姆霍兹自由能的变化 $\Delta F_{brick}$ (或微小变化 $dF_{brick}$) 与最大可提取功 $W_{by}$ (或 $W_{max}$) 的关系是: $$ W_{by} = -(U_{brick, final} - U_{brick, initial}) + T_{env}(S_{brick, final} - S_{brick, initial}) $$ $$ W_{by} = -( (U_{brick, final} - T_{env}S_{brick, final}) - (U_{brick, initial} - T_{env}S_{brick, initial}) ) $$ $$ W_{by} = -(F_{brick, final} - F_{brick, initial}) = -\Delta F_{brick} $$ 或者写成微分形式 (假设卡诺效率): $$ W_{by} = -dF_{brick} $$
- 一般情况下,实际做的功会小于这个最大值,即 $W_{by} \le -dF_{brick}$。
- $\Delta F$ 告诉我们系统在等温过程中 (与温度为 $T_{env}$ 的热源接触) 最多可以提取多少功。
- 技术比喻:你可以把亥姆霍兹自由能想象成系统在特定环境温度下“储存”的能够转化为功的“能量潜力”。当系统从一个非平衡态向平衡态转变时,自由能的减少量就等于它能对外做的最大功。就像一个物体在高处拥有重力势能,当它下落到最低点 (平衡态) 时,势能可以转化为其他形式的能量 (功)。亥姆霍兹自由能也类似,系统偏离平衡态时具有较高的自由能,自发趋向平衡态 (自由能最低点) 的过程中,可以对外做功。
3. 自由能与平衡 (Free Energy and Equilibrium)
-
练习 (Exercise, Slides 10, 11):
- 讲义中计算了一个砖块在 $T_{env}=300\text{K}$ 的环境下,从 $300\text{K}$ 被加热到 $310\text{K}$ ( $\Delta T = +10\text{K}$ ) 和被冷却到 $290\text{K}$ ( $\Delta T = -10\text{K}$ ) 时的亥姆霍兹自由能变化 $\Delta F_B$。
- 使用的公式是: $\Delta U_B = C \Delta T = C(T_B - 300\text{K})$ $\Delta S_B = C \ln(T_B / 300\text{K})$ $\Delta F_B = \Delta U_B - (300\text{K}) \Delta S_B$
- 计算结果显示,无论是加热还是冷却,$\Delta F_B$ 都是正的 (加热时 $0.16 \text{ kJ}$,冷却时 $0.17 \text{ kJ}$)。
- 这说明当砖块的温度 $T_B$ 等于环境温度 $T_{env}$ ($300\text{K}$) 时,其亥姆霍兹自由能处于最小值。
- 图像也直观地展示了这一点:$F_B$ 关于 $T_B$ 的曲线在 $T_B = T_{env} = 300\text{K}$ 处有极小值。
-
平衡态对应自由能最小 (Equilibrium corresponds to Free Energy Minimum):
- 我们知道,一个孤立系统达到平衡时,其总熵 $S_{tot}$ 达到最大值。
- 现在考虑一个系统 (sys) 与一个大的恒温环境 (e, environment or reservoir) 接触,环境温度为 $T_e$。总熵 $S_{tot} = S_{sys} + S_e$。
- 如果系统从环境中吸收了微小的内能 $dU$ 。那么环境的熵变 $dS_e = dU_e/T_e = -dU_{sys}/T_e$。
- 总熵的微小变化为: $$ dS_{tot} = dS_{sys} + dS_e = dS_{sys} - \frac{dU_{sys}}{T_e} $$
- 我们可以整理这个式子: $$ dS_{tot} = - \frac{(dU_{sys} - T_e dS_{sys})}{T_e} $$
- 我们定义的亥姆霍兹自由能 $F_{sys} = U_{sys} - T_e S_{sys}$ (注意这里的 $T_e$ 是常数,是环境的温度)。其微分形式为 $dF_{sys} = dU_{sys} - T_e dS_{sys} - S_{sys}dT_e$。如果在恒温条件下 ($dT_e=0$),或者更准确地说,我们定义 $F$ 时用的 $T$ 就是环境温度 $T_e$ (一个常数),那么对于系统的变化: $$ dF_{sys} (\text{at constant } T_e) = dU_{sys} - T_e dS_{sys} $$
- 所以: $$ dS_{tot} = - \frac{dF_{sys}}{T_e} $$
- 这个关系非常重要!它告诉我们:
- 在一个与恒温热源接触的系统中 (保持恒容),总熵 $S_{tot}$ 的最大化等价于系统亥姆霍兹自由能 $F_{sys}$ 的最小化。
- 因为 $T_e$ 是正的,所以 $dS_{tot}$ 和 $dF_{sys}$ 符号相反。当系统趋于平衡 ($dS_{tot} > 0$ 或 $dS_{tot} = 0$ 达到最大值) 时,$dF_{sys} < 0$ 或 $dF_{sys} = 0$ (达到最小值)。
- 因此,在恒温恒容条件下,系统达到平衡态的判据是其亥姆霍兹自由能达到最小值。
- Slide 12右下角的图示也形象地表明了 $F = U - TS$ 的关系。$F$ 的极小值点对应平衡态。
三、亥姆霍兹自由能的重要性及使用条件
1. 重要性 (Importance)
- 简化平衡判据: 在很多情况下 (特别是恒温恒容),最大化总熵 $S_{tot}$ (有时计算复杂) 来判断平衡,可以等效地用最小化系统自由能 $F_{sys}$ (通常更容易计算) 来判断。
- 衡量可做功: 当系统偏离平衡态时,其多余的自由能 (excess free energy) $F - F_{equilibrium}$ 表示一个理想热机在该环境下能从系统中提取的最大功。
- 数据可查: 很多材料的自由能数据是可以查表得到的 (例如化学燃料)。
- 后续应用: 亥姆霍兹自由能是后续学习化学势 (Chemical potential)、半导体中载流子浓度、表面吸附、相变等高级内容的基础。
2. 使用亥姆霍兹自由能的条件 (Conditions for using Helmholtz Free Energy)
使用亥姆霍兹自由能作为平衡判据 (即平衡时 $F_{sys}$ 最小) 或计算最大可做功 ($W_{max} = -\Delta F$) 是有条件的:
- 系统与大环境接触: 系统与一个大的环境 (热储, thermal reservoir) 相连,环境温度恒为 $T_{env}$。
-
只有热量交换: 系统与环境之间只有热量可以流动。这意味着系统的体积 (Volume, $V$) 保持不变,粒子数 (Particle number, $N$) 也保持不变。
- 简而言之,亥姆霍兹自由能适用于恒温 (isothermal)、恒容 (isochoric)、恒定粒子数的过程。
3. 注意事项:与吉布斯自由能的区别 (Caution: Difference from Gibbs Free Energy)
-
Maximum $S_{tot}$ Does Not Always Mean Minimum $F_{sys}$ (Helmholtz):
- 我们在推导 $F_{brick} = U_{brick} - T_{env}S_{brick}$ 时,隐含了一个假设,即砖块的体积是恒定的。
- 如果体积不是恒定的 (例如,系统是一个在恒定大气压下的气体),那么系统可以通过膨胀或收缩来做功或被做功,这会改变分析。
- 在更常见的情况下,系统经历的是恒压 (isobaric) 而非恒容过程。这时,我们需要使用另一个自由能,叫做吉布斯自由能 (Gibbs Free Energy, $G$): $$ G = U + pV - TS = H - TS $$ 其中 $H = U + pV$ 是焓 (Enthalpy)。$pV$ 这一项考虑了系统在体积变化时所做的功。
- 亥姆霍兹自由能主要用于恒容过程,而吉布斯自由能用于恒压过程。选择哪种自由能取决于过程的约束条件。
例题
题目:一个热砖块 $T_{brick} = 400 \text{ K}$,热容 $C = 1 \text{ J/K}$,与一个 $T_{res} = 300 \text{ K}$ 的热储接触,通过一个引擎对外做功。问能从砖块中提取的最大功是多少?
这个问题就是计算砖块从 $400 \text{ K}$ 冷却到与热储相同的温度 $300 \text{ K}$ 过程中,亥姆霍兹自由能的变化的负值。 环境温度 $T_{env} = T_{res} = 300 \text{ K}$。 初始状态:$T_i = 400 \text{ K}$ 末状态:$T_f = 300 \text{ K}$
- 计算内能变化 $\Delta U_{brick}$: $$ \Delta U_{brick} = \int_{T_i}^{T_f} C dT = C (T_f - T_i) = (1 \text{ J/K}) (300 \text{ K} - 400 \text{ K}) = -100 \text{ J} $$
- 计算熵变 $\Delta S_{brick}$: $$ \Delta S_{brick} = \int_{T_i}^{T_f} \frac{C}{T} dT = C \ln\left(\frac{T_f}{T_i}\right) = (1 \text{ J/K}) \ln\left(\frac{300}{400}\right) = (1 \text{ J/K}) \ln(0.75) $$ $\ln(0.75) \approx -0.2877$ $$ \Delta S_{brick} \approx -0.2877 \text{ J/K} $$
- 计算最大可提取功 $W_{max}$: $$ W_{max} = -\Delta F_{brick} = -(\Delta U_{brick} - T_{env} \Delta S_{brick}) = -\Delta U_{brick} + T_{env} \Delta S_{brick} $$ $$ W_{max} = -(-100 \text{ J}) + (300 \text{ K}) (-0.2877 \text{ J/K}) $$ $$ W_{max} = 100 \text{ J} - 300 \times 0.2877 \text{ J} = 100 \text{ J} - 86.31 \text{ J} = 13.69 \text{ J} $$ 所以,最大可提取功是 $13.69 \text{ J}$。
好的同学,我们继续学习 Lecture 10 的内容,这次课的主题是平衡与化学势 (Equilibrium and Chemical Potential)。这一讲在上一讲亥姆霍兹自由能的基础上,引入了化学势的概念,用来处理有粒子数交换或化学反应的系统平衡问题。
Lecture 10: 平衡与化学势 (Equilibrium and Chemical Potential)
从熵最大化到自由能最小化:引入粒子交换
1. 以往的平衡判据 (Previously…Maximize entropy to find equilibrium)
- 我们之前学习过,对于一个孤立系统,或几个可以相互作用的系统组成的孤立整体,它们达到平衡的条件是总熵最大化 ($dS_{tot} = 0$)。
- 例如,两个可以交换体积 (Volume) 的系统 (System 1, System 2): 当 $dS_{tot}/dV_1 = 0 \implies dS_1/dV_1 = dS_2/dV_2 \implies p_1/T_1 = p_2/T_2$ (如果同时可以交换能量使得 $T_1=T_2$) $\implies p_1 = p_2$。 更准确地,讲义中从 $(\partial S_1/\partial V_1){U_1,N_1} = (\partial S_2/\partial V_2){U_2,N_2}$ (假设 $dV_1 = -dV_2$),并利用 $TdS = dU + pdV \implies (\partial S/\partial V)_{U,N} = p/T$。所以平衡时 $p_1/T_1 = p_2/T_2$。如果它们也处于热平衡 $T_1=T_2$,则 $p_1=p_2$。 讲义中直接给出了 $(\partial S_1/\partial V_1) = N_1/V_1$ 和 $(\partial S_2/\partial V_2) = N_2/V_2$ (这实际上是理想气体在特定条件下的简化结果,更普适的是 $p/T$)。
-
两个可以交换能量 (Energy) 的系统: 当 $dS_{tot}/dU_1 = 0 \implies dS_1/dU_1 = dS_2/dU_2 \implies 1/T_1 = 1/T_2 \implies T_1 = T_2$。 这里 $(\partial S/\partial U)_{V,N} = 1/T$。
- 现在的新情况: 允许系统间交换粒子数 (Particle numbers)。
2. 自由能、平衡与化学势 (Free Energy, Equilibrium and Chemical Potential)
- 当系统与一个大的恒温热储 (reservoir at temperature $T_{reservoir}$) 接触时,我们已经知道平衡条件是最大化总熵 ($S_{tot} = S_{reservoir} + S_{small_system}$),这等价于最小化小系统的亥姆霍兹自由能 ($F_{sys} = U_{sys} - T_{reservoir}S_{sys}$)。
- 为什么等价? 当系统从热储吸收能量 $dU_{sys}$ 时,热储能量减少 $dU_{reservoir} = -dU_{sys}$,热储熵变 $dS_{reservoir} = -dU_{sys}/T_{reservoir}$。总熵变 $dS_{tot} = dS_{sys} + dS_{reservoir} = dS_{sys} - dU_{sys}/T_{reservoir} = -(dU_{sys} - T_{reservoir}dS_{sys})/T_{reservoir} = -dF_{sys}/T_{reservoir}$。所以 $S_{tot}$ 最大化对应 $F_{sys}$ 最小化。
- 使用自由能的好处是,我们只需要关注系统本身的性质和热储的温度,而不需要显式计算热储的熵。
-
考虑两个子系统 (1 和 2) 之间交换粒子:
- 这两个子系统都与一个共同恒温热储接触,温度为 $T$。
- 总的自由能 $F_{tot} = F_1 + F_2$。
- 当粒子从系统2转移到系统1时,$dN_1 > 0$ 且 $dN_2 = -dN_1 < 0$。
- 在平衡时,总自由能对于粒子数的微小变化应该达到最小值,即 $dF_{tot}/dN_1 = 0$。 $$ \frac{dF_{tot}}{dN_1} = \frac{d(F_1 + F_2)}{dN_1} = \frac{dF_1}{dN_1} + \frac{dF_2}{dN_1} $$ 由于 $dN_2 = -dN_1$,所以 $dF_2/dN_1 = (dF_2/dN_2)(dN_2/dN_1) = -dF_2/dN_2$。 因此,平衡条件为: $$ \frac{dF_1}{dN_1} - \frac{dF_2}{dN_2} = 0 \implies \frac{dF_1}{dN_1} = \frac{dF_2}{dN_2} $$
-
化学势 (Chemical Potential, $\mu$) 的定义: 亥姆霍兹自由能对粒子数的变化率被定义为一个非常重要的量,称为化学势: $$ \mu_i \equiv \left(\frac{\partial F_i}{\partial N_i}\right)_{T,V} $$ (下标 $T,V$ 表示在恒温恒容条件下求偏导)。
- 粒子交换的平衡条件: 对于两个交换粒子的子系统,达到平衡时,它们的化学势相等: $$ \mu_1 = \mu_2 $$
- 总结一下平衡的演进: 最大化总熵 $\rightarrow$ 最小化自由能 (对于与热储接触的系统) $\rightarrow$ 化学势相等 (对于可交换粒子的系统)。
化学势的作用
1. 统一平衡条件 (Unifying Equilibrium Conditions)
- 引入化学势使得各种平衡条件在形式上统一起来:
- 交换体积 (Volume): $(\partial S_1/\partial V_1) = (\partial S_2/\partial V_2)$ (若用熵) $\implies p_1 = p_2$ (若$T_1=T_2$)
- 交换能量 (Energy): $(\partial S_1/\partial U_1) = (\partial S_2/\partial U_2)$ (若用熵) $\implies T_1 = T_2$
- 交换粒子 (Particles): $(\partial F_1/\partial N_1) = (\partial F_2/\partial N_2)$ (若用自由能) $\implies \mu_1 = \mu_2$
- 为什么粒子交换用 $dF/dN$ 而不是 $dS/dN$? 当粒子在两个子系统间交换时,如果这两个子系统还与一个共同的外部热储相连,那么粒子移动时可能伴随着能量的吸收或释放,这会影响热储的熵。亥姆霍兹自由能 $F = U - TS_{sys}$ (这里的 $T$ 是热储的温度) 已经内含了与热储在恒温下进行热交换的效应。因此,最小化系统的自由能 $F_{sys}$ 是在有热储存在时正确的平衡判据。
2. 粒子流动的方向 (Direction of Particle Flow)
- 当系统未达到平衡时 (例如 $\mu_1 > \mu_2$):
- 粒子会自发地从化学势高的地方流向化学势低的地方,以使得总自由能 $F_{tot}$ 最小化。
- 这与能量从高温流向低温,或物体从高压区移向低压区是类似的。
- Slide 14 的自由膨胀例子:初始时,左边气体密度高 (化学势高),右边真空 (化学势低)。移除隔板后,粒子从左向右扩散,直到两边化学势相等 (密度均匀)。
3. 化学势的物理解释 (Physical Interpretation)
(Slide 10)
- 化学势 $\mu$ 可以近似理解为 (在恒温恒容下) 向系统中加入一个粒子所引起的自由能的变化: $$ \mu \approx \frac{F(N+1) - F(N)}{1} \quad (\text{at constant } T, V) $$
四、计算化学势 (Computing $\mu$)
1. Example
- 题目:系统在恒定温度 $T$ 和体积 $V$ 下,内能 $U(N) = aN^2$,熵 $S(N) = cN$。求化学势 $\mu$。
- 解答:
- 首先计算亥姆霍兹自由能 $F(N) = U(N) - TS(N)$: $$ F(N) = aN^2 - T(cN) = aN^2 - cTN $$
- 然后根据定义计算化学势 $\mu = dF/dN$:
$$
\mu = \frac{dF}{dN} = \frac{d(aN^2 - cTN)}{dN} = 2aN - cT
$$
- 所以正确答案是 (d) $2aN - cT$。
2. 理想气体的化学势 (Chemical Potential for an Ideal Gas)
- 对于理想气体,单位粒子的内能 $u = U/N$ 仅取决于温度 $T$,与 $N$ 无关。很多情况下可以设 $u=0$。
- 熵 $S$ 与 $N$ 的关系: 考虑 $N$ 个粒子分布在 $M$ 个”箱子” (bins) 或量子态中。微观状态数 $\Omega = M^N/N!$ (假设粒子可分辨且每个箱子可容纳多个粒子,或者粒子不可分辨但大多箱子为空。更准确的玻尔兹曼统计的 Sackur-Tetrode 方程会更复杂,这里是简化模型)。 $$ S = k \ln \Omega = k \ln\left(\frac{M^N}{N!}\right) = k(N \ln M - \ln N!) $$ 其中 $k$ 是玻尔兹曼常数。
- 计算 $(\partial S/\partial N){M,T}$: 使用斯特林近似 (Stirling’s Approximation) $\ln N! \approx N \ln N - N$,则 $d(\ln N!)/dN \approx \ln N$。 (更准确地,$(\partial S/\partial N){M,T} = k (\ln M - (\ln N - 1) -1) = k(\ln M - \ln N) = k \ln(M/N)$ for $N \gg 1$) 讲义中给出 $(\partial S/\partial N)_M = k \ln(M/N)$ (这里 $M$ 是总的可用状态数,与体积 $V$ 和温度 $T$ 有关)。
- 化学势 $\mu$: $$ \mu = \left(\frac{\partial F}{\partial N}\right){T,V} = \left(\frac{\partial (U-TS)}{\partial N}\right){T,V} = u - T\left(\frac{\partial S}{\partial N}\right){T,V} $$ 代入 $(\partial S/\partial N){T,V} = k \ln(M/N)$: $$ \mu = u - kT \ln\left(\frac{M}{N}\right) = u + kT \ln\left(\frac{N}{M}\right) $$
- 引入粒子数密度 (particle density) $n = N/V$ 和单位体积量子态数 (number of states per unit volume) $n_Q = M/V$ (也称为量子浓度, quantum concentration,它依赖于温度 $T$ 和粒子质量)。
则 $N/M = (N/V) / (M/V) = n/n_Q$。
所以理想气体的化学势为:
$$
\mu = u + kT \ln\left(\frac{n}{n_Q}\right)
$$
- 重要结论: 对于理想气体,化学势与粒子数密度的对数成正比。密度越高,化学势越大。
- 如果将单粒子内能 $u$ 设为能量零点 (例如对于某些问题可以令 $u=0$),则 $\mu = kT \ln(n/n_Q)$。
五、化学势的应用实例
1. 固溶体中的缺陷 (Solid “Solutions” / Defects in Solids)
- 考虑一个模型:Si 晶体中有 $M$ 个可能的”Si”格点位,其中 $N$ 个位置被”Au”原子占据($N \ll M$)。这可以看作是 Au 在 Si 中的固溶。
- 能量: 每当一个 Au 原子进入 Si 格点时,系统能量增加 $\Delta$ (相对于 Au 原子在纯 Au 晶体中)。所以总能量 $U(N) = N\Delta$ (选择纯 Au 的能量为零点)。
- 熵: 熵主要来自 Au 原子在 $M$ 个格点上的排布方式 (构型熵, configurational entropy),忽略振动等其他贡献。 微观状态数 $\Omega(N) = \binom{M}{N} = \frac{M!}{N!(M-N)!}$。 熵 $S(N) = k \ln \Omega(N) = k \ln \frac{M!}{N!(M-N)!}$。
- 自由能: $F(N) = U(N) - TS(N) = N\Delta - kT \ln \frac{M!}{N!(M-N)!}$。
-
平衡条件: 在平衡时,Si 中 Au 的化学势应该等于纯 Au 中 Au 的化学势。如果我们方便地将纯 Au 中 Au 的化学势设为零 (能量零点选择),那么 Si 中 Au 的化学势在平衡时也应为零。即 $(\partial F/\partial N){T,V} = 0$。
$$
\mu = \left(\frac{\partial F}{\partial N}\right){T,V} = \left(\frac{\partial U}{\partial N}\right){T,V} - T\left(\frac{\partial S}{\partial N}\right){T,V} = 0
$$
- $(\partial U/\partial N)_{T,V} = \Delta$。
- 使用斯特林近似,对于 $N \ll M$ 的情况: $S(N) \approx k [M \ln M - M - (N \ln N - N) - ((M-N)\ln(M-N) - (M-N))]$ $(\partial S/\partial N)_{T,V} \approx k [-\ln N + \ln(M-N)] = k \ln\left(\frac{M-N}{N}\right) \approx k \ln\left(\frac{M}{N}\right)$ (因为 $N \ll M$)。 (Slide 18 的推导中用了 $d(\ln X!)/dX \approx \ln X$,这是斯特林近似 $\ln X! \approx X \ln X - X$ 的微分结果。)
- 所以化学势为: $$ \mu = \Delta - kT \ln\left(\frac{M}{N}\right) $$
- 令 $\mu = 0$ 求平衡时的缺陷浓度 $N/M$: $$ \Delta - kT \ln\left(\frac{M}{N}\right) = 0 \implies kT \ln\left(\frac{M}{N}\right) = \Delta $$ $$ \ln\left(\frac{M}{N}\right) = \frac{\Delta}{kT} \implies \frac{M}{N} = e^{\Delta/kT} $$ 所以,平衡时缺陷 (或溶质原子) 的分数浓度为: $$ \frac{N}{M} = e^{-\Delta/kT} $$ 这正是玻尔兹曼因子 (Boltzmann factor) 的形式!这意味着在一定温度下,形成一个能量为 $\Delta$ 的缺陷的概率与 $e^{-\Delta/kT}$ 成正比。 也可以表示为缺陷密度 $n = N/V$ 与格点密度 $n_c = M/V$ 的比: $$ \frac{n}{n_c} = e^{-\Delta/kT} $$
- Checkpoint 3 (Sugar in water): 糖在水中的溶解度也遵循类似 $N_{sugar}/N_W \propto e^{-\Delta/kT}$ 的关系 (这里 $\Delta$ 是溶解能)。要使溶解的糖最多,即 $N_{sugar}$ 最大,需要最高的温度 $T$。所以选 (c) Near boiling。
- Arrhenius Plot (阿伦尼乌斯图): 如果将 $\ln(\text{concentration})$ 对 $1/T$ 作图,会得到一条直线,其斜率为 $-\Delta/k$。这种图常用于从实验数据中提取活化能或能量差。图上显示 Cr 的斜率更陡 (负得更多),说明 Cr 的 $\Delta/k$ 更大,即 Cr 在硅中的能量成本更高。
2. 本征半导体中的电子和空穴 (Electrons and Holes in Intrinsic Semiconductors)
- 能带结构 (Band Structure): 半导体中电子的能量不是任意的,存在允带和禁带。低能量区为价带 (valence band),高能量区为导带 (conduction band),它们之间有一个能量间隔,称为禁带宽度 (energy gap, $\Delta$ 或 $E_g$)。
-
电子-空穴对 (Electron-hole pairs):
- 在 $T=0\text{K}$ 时,价带被电子填满,导带为空,没有自由载流子。
- 当 $T > 0\text{K}$ 时,部分电子从价带受热激发到导带,成为自由移动的导电电子 (conduction electrons)。
- 电子离开价带后,在价带中留下一个空的量子态,称为空穴 (hole)。空穴可以被看作是带正电荷的准粒子,也能自由移动。
- 在本征半导体 (Intrinsic Semiconductor) 中,电子和空穴是成对产生的,所以电子浓度 $n_e$ 等于空穴浓度 $n_h$。我们称之为本征载流子浓度 $n_i = n_e = n_h$。
- 平衡条件: 导电电子和空穴可以看作是两种理想气体。系统总自由能 $F = F_e + F_h$。当电子-空穴对产生或复合达到平衡时,系统自由能最小。 由于电子和空穴是成对产生的 ($dN_e = dN_h$),所以 $dF/dN_e = (\partial F_e/\partial N_e) + (\partial F_h/\partial N_h) = 0$。 这意味着在平衡时: $$ \mu_e + \mu_h = 0 $$ 其中 $\mu_e$ 是导带电子的化学势,$\mu_h$ 是价带空穴的化学势。
-
计算本征载流子浓度 $n_i$ :
- 取价带顶的能量为能量零点。则空穴的单粒子内能 $u_h \approx 0$。
- 导带底的能量为 $\Delta$。则电子的单粒子内能 $u_e \approx \Delta$。
- 根据理想气体化学势公式: $\mu_h = kT \ln(n_h/n_{Qh})$ $\mu_e = \Delta + kT \ln(n_e/n_{Qe})$ (其中 $n_{Qe}$ 和 $n_{Qh}$ 分别是电子和空穴的量子浓度)。
- 代入 $\mu_e + \mu_h = 0$: $$ \Delta + kT \ln\left(\frac{n_e}{n_{Qe}}\right) + kT \ln\left(\frac{n_h}{n_{Qh}}\right) = 0 $$
- 由于 $n_e = n_h = n_i$: $$ \Delta + kT \ln\left(\frac{n_i^2}{n_{Qe}n_{Qh}}\right) = 0 $$ $$ \ln\left(\frac{n_i^2}{n_{Qe}n_{Qh}}\right) = -\frac{\Delta}{kT} $$ $$ n_i^2 = n_{Qe}n_{Qh} e^{-\Delta/kT} $$
- 所以本征载流子浓度为: $$ n_i = \sqrt{n_{Qe}n_{Qh}} e^{-\Delta/(2kT)} $$ 如果定义一个等效的量子浓度 $n_Q = \sqrt{n_{Qe}n_{Qh}}$ (有时也写作 $N_C N_V$ 的形式,其中 $N_C, N_V$ 是导带和价带的有效状态密度),则: $$ n_i = n_Q e^{-\Delta/(2kT)} $$
- 这个结果表明,本征载流子浓度随温度指数增长,且强烈依赖于禁带宽度 $\Delta$。$\Delta$ 越大,$n_i$ 越小。
- Slide 28 的问题: $n_Q$ 的值对于不同材料不同,且不简单等于用电子质量 $m_e$ 计算的值,是因为电子和空穴在晶格中运动时,受到晶格势场的作用,表现出有效质量 (effective mass) $m_{e,effective}$ 和 $m_{h,effective}$,它们通常不等于自由电子质量,且彼此也不同。因此 $n_Q$ 通常作为经验参数处理。
- Slide 30 的 Checkpoint: 要使导电电子数与总电子数的比例 $N_c/N$ (近似于 $n_i/N_{total_valence_electrons}$) 最大,需要 $n_i$ 最大。根据 $n_i \propto e^{-\Delta/(2kT)}$,在相同温度 $T$ 下,$n_i$ 最大对应于禁带宽度 $\Delta$ 最小的材料。表格中 PbS 的 $\Delta = 0.37 \text{ eV}$ 最小。所以选 (e) PbS。
3. 数字温度计 (Digital Thermometers)
- 半导体材料 (如热敏电阻, thermistor) 的电阻率强烈依赖于温度 (因为载流子浓度 $n_i$ 随温度指数变化)。这种电阻随温度的快速 (指数级) 变化特性被用于制造数字温度计。
由非平衡化学势做功 (Work from Free Energy Due to Non-equilibrium $\mu$)
- 考虑 $N$ 个粒子在温度 $T$ 下的等温膨胀。我们知道系统做的功 $W_{by} = NkT \ln(V_f/V_i)$。
- 这也可以从自由能变化的角度来看:$W_{by} = -\Delta F = -(\Delta U - T\Delta S)$。
- 对于理想气体的等温膨胀,$\Delta U = 0$ (内能只与温度有关)。所以 $W_{by} = T\Delta S$。
- 这表明,即使是旧的物理过程 (如理想气体等温膨胀),也可以用新的语言 (自由能、化学势) 来描述。初始时,如果气体被限制在 $V_i$,然后膨胀到 $V_f$,可以看作是由于初始(如果想象有隔板)和最终状态之间存在化学势(或压强,与化学势相关)的非平衡,系统对外做功。
Lecture 11: Gibs Free Energy
核心概念梳理
1. 数学准备:微分 (Differentials)
- 应用到热力学函数: 比如亥姆霍兹自由能 (Helmholtz Free Energy) $F = U - TS$ (Slide 5)。它的微分是: $$ dF = dU - d(TS) $$ 应用乘法法则于 $d(TS)$: $$ d(TS) = TdS + SdT $$ 所以: $$ dF = dU - TdS - SdT $$
- 计算有限变化: 从初始状态 $i$ 到最终状态 $e$ 的有限变化 $\Delta g$ 可以通过对微分 $dg$ 积分得到 (Slide 6)。如果 $f = \frac{dg}{dh}$,那么 $dg = f dh$。要计算 $\Delta g$,我们需要对 $f dh$ 进行积分: $$ \Delta g = \int_{h(f_i)}^{h(f_e)} f dh $$ 注意积分上下限是对应于 $f_i$ 和 $f_e$ 的 $h$ 的值。
2. 热力学基本关系 (Fundamental Relation of Thermodynamics)
(Slides 2, 15, 18, 28)
这是热力学中一个非常核心的方程,它联系了内能 (Internal Energy) $U$、熵 (Entropy) $S$、体积 (Volume) $V$ 和粒子数 (Particle Number) $N$ 的变化。
- 熵的微分形式 (Slide 15): $$ dS = \left(\frac{\partial S}{\partial U}\right){V,N} dU + \left(\frac{\partial S}{\partial V}\right){U,N} dV + \left(\frac{\partial S}{\partial N}\right){U,V} dN $$ 利用热力学关系 $1/T = (\partial S/\partial U){V,N}$, $p/T = (\partial S/\partial V){U,N}$, 和 $-\mu/T = (\partial S/\partial N){U,V}$ (其中 $\mu$ 是化学势 Chemical Potential),我们可以得到: $$ dS = \frac{1}{T}dU + \frac{p}{T}dV - \frac{\mu}{T}dN $$
-
内能的微分形式 (Slide 18, 20, 28):
更常见的形式是表示 $dU$:
$$
dU = TdS - pdV + \mu dN
$$
这个方程适用于任何准静态过程 (quasi-static process) 的简单系统。
- 把这个方程想象成描述一个系统能量“预算”的公式。当系统的熵、体积或粒子数发生微小变化时,其内能会如何相应地改变。$TdS$ 是热量交换项,$-pdV$ 是体积功项,$\mu dN$ 是与粒子数变化相关的化学功。
3. 亥姆霍兹自由能 (Helmholtz Free Energy, F)
(Slides 5, 12, 20, 21, 28)
- 定义 (Slide 12, 20): $$ F = U - TS $$
- 微分形式 (Slide 20): 从 $dF = dU - TdS - SdT$,代入 $dU = TdS - pdV + \mu dN$: $$ dF = (TdS - pdV + \mu dN) - TdS - SdT $$ $$ dF = -SdT - pdV + \mu dN $$
-
适用条件与意义:
- 当系统与恒温热源接触,且体积保持不变时 (即 $T$ 和 $V$ 恒定),亥姆霍兹自由能的变化 $\Delta F$ 可以判断过程的自发性。
- 在恒温恒容 ($dT=0, dV=0$) 条件下,一个自发过程会使得系统的亥姆霍兹自由能减小 ($\Delta F \le 0$)。
- 平衡态时,$F$ 达到最小值 ($dF = 0$) (Slide 21)。
- $W_{max} = -\Delta F$ 代表恒温可逆过程中系统对外做的最大功。
4. 吉布斯自由能 (Gibbs Free Energy, G)
(Slides 2, 11, 12, 13, 18, 20, 22, 23, 25, 28)
-
引入原因 (Slide 11): 在很多实际情况下,尤其是化学反应,系统通常与恒温恒压的环境接触 (例如,在敞口烧杯中进行的反应,压力为大气压,温度为室温)。亥姆霍兹自由能适用于恒容过程,而对于恒压过程,我们需要一个新的热力学势——吉布斯自由能。 $pV$ 项考虑了在体积变化过程中系统对环境做的功。
-
定义 (Slide 11, 12, 20): $$ G = U + pV - TS $$ 也可以写成 $G = H - TS$ (其中 $H = U + pV$ 是焓 Enthalpy),或者 $G = F + pV$。
- 微分形式 (Slide 18, 20): 从 $G = F + pV$ 出发,$dG = dF + d(pV) = dF + pdV + Vdp$。 代入 $dF = -SdT - pdV + \mu dN$: $$ dG = (-SdT - pdV + \mu dN) + pdV + Vdp $$ $$ dG = -SdT + Vdp + \mu dN $$
-
适用条件与意义 (Slide 2, 13, 22, 23):
- 当系统与恒温恒压环境接触时 (即 $T$ 和 $P$ 恒定),吉布斯自由能的变化 $\Delta G$ 可以判断过程的自发性。
- 在恒温恒压 ($dT=0, dp=0$) 条件下:
- $\Delta G < 0$: 过程自发进行 (spontaneous)。
- $\Delta G > 0$: 过程非自发 (non-spontaneous),需要外界做功才能发生。
- $\Delta G = 0$: 系统处于平衡态 (equilibrium)。
- 平衡态时,$G$ 达到最小值 ($dG=0$)。
- $W_{useful, max} = -\Delta G$ 代表恒温恒压可逆过程中系统对外做的最大非体积功 (例如电功)。
5. 化学势 (Chemical Potential, μ)
(Slides 7, 8, 10, 17, 18, 19, 24, 25, 28)
- 定义 (Slide 20, 28): 化学势是当其他条件 (如 $S,V$ 或 $T,V$ 或 $T,P$) 不变时,向系统中加入一个粒子所引起的能量 (如 $U, F, G$) 的变化。 $$ \mu = \left(\frac{\partial U}{\partial N}\right){S,V} = \left(\frac{\partial F}{\partial N}\right){T,V} = \left(\frac{\partial G}{\partial N}\right)_{T,P} $$
-
物理意义:
- 衡量粒子“逃逸”或“进入”系统的趋势。如果一个区域的化学势高,粒子倾向于离开该区域。
- 在多相或多组分系统中,当达到平衡时,各相或各组分之间可自由交换的粒子的化学势相等。
- 技术比喻: 化学势类似于物质的“浓度压力”或“化学压力”。物质会自发地从化学势高的地方流向化学势低的地方,直到各处化学势相等,达到平衡。就像温度决定热量流动方向,压力决定气体流动方向一样。
-
$G = \mu N$ (Slides 19, 25, 28):
- 对于恒定 $T, P$ 下的单组分系统,吉布斯自由能可以简单表示为 $G = \mu N$。这是因为 $G$ 是广延量 (extensive property),$\mu$ 是强度量 (intensive property)。
- Slide 19 通过理想气体的例子说明,在恒温恒压下,两个相连的容器,尽管粒子数 $N_A, N_B$ 可以不同,但平衡时密度相同,化学势 $\mu_A = \mu_B$,且这个化学势不依赖于容器中有多少粒子。
- Slide 25 指出,当有多种相态 (气、液、固) 共存时,为了使总的 $G = \mu_{gas}N_{gas} + \mu_{liquid}N_{liquid} + \mu_{solid}N_{solid}$ 最小化,粒子会倾向于聚集在化学势最低的那个相态中。
- 在给定压强与温度下,物质的固态、液态以及气态的密度是固定的。添加更多的固体、液体或者气体只会改变该物质的体积,不会改变密度与化学势。化学势与粒子数无关
应用与实例
1. 化学平衡 (Chemical Equilibrium)
(Slide 7) 对于一个化学反应 $aA + bB \leftrightarrow cC$:
- 在平衡时,总的自由能 ($F$ 或 $G$,取决于条件) 达到最小值,因此 $dF=0$ 或 $dG=0$。
- 对于恒温恒容条件 (使用 $F$) 或恒温恒压条件 (使用 $G$),可以导出平衡条件: $$ a\mu_A + b\mu_B = c\mu_C $$ 其中 $\mu_A, \mu_B, \mu_C$ 分别是组分 A, B, C 的化学势。
2. 大气定律 (The Law of Atmospheres)
(Slide 8)
- 考虑不同高度的两部分气体,它们可以交换粒子,整个大气是热源。
- 平衡时,两部分气体的化学势相等: $\mu_1 = \mu_2$。
- 对于理想气体,化学势表达式为 $\mu = kT \ln(n/n_Q) + U_{ext}$,其中 $U_{ext}$ 是外部势能。这里 $U_{ext,1}=0$, $U_{ext,2}=mgh$。
- $\mu_1 = kT \ln(n_1/n_Q)$
- $\mu_2 = kT \ln(n_2/n_Q) + mgh$
- 令 $\mu_1 = \mu_2$,可以解出粒子数密度 $n_2/n_1 = e^{-mgh/kT}$。
- 结合理想气体状态方程 $p=nkT$ (温度 $T_1=T_2$ 处于平衡),得到 $p_2/p_1 = e^{-mgh/kT}$。
3. 固-气平衡:蒸汽压 (Solid-gas equilibrium: vapor pressure)
(Slide 10)
- 考虑固相和气相在恒定体积和温度下达到平衡 (例如,密闭容器中的干冰 $CO_2$)。
- 此时应使用亥姆霍兹自由能 $F$。但通常化学势的讨论更直接。
- 平衡条件是固相的化学势等于气相的化学势: $\mu_s = \mu_g$。
- 对于固体,如果忽略其熵,$\mu_s \approx - \Delta$ (其中 $\Delta$ 是将一个原子从固体中移到真空中所需的能量,即结合能)。
- 对于理想气体,$\mu_g = kT \ln(n/n_Q) = kT \ln(p/p_Q)$ (其中 $n_Q$ 是量子浓度, $p_Q = n_Q kT$)。
- 令 $\mu_s = \mu_g$: $$ kT \ln(p/p_Q) = -\Delta $$ $$ p = p_Q e^{-\Delta/kT} $$ 这个 $p$ 就是该温度下的饱和蒸汽压 (vapor pressure) $p_{vapor}$。
- 如果实际气体压强 $p < p_{vapor}$,固体会升华 (sublimation) 或液体会蒸发 (evaporation),直到气压达到 $p_{vapor}$。
4. 水的结冰自发性 (Spontaneity of Water Freezing)
(Slide 14)
- 这是一个判断过程自发性的例子,通常在恒压下发生 (大气压),所以使用吉布斯自由能。
- 给定 $\Delta H$ (焓变) 和 $\Delta S$ (熵变) 以及温度 $T$。
- 计算 $\Delta G = \Delta H - T\Delta S$。
- 如果 $\Delta G < 0$,在给定温度下结冰是自发的。
- 如果 $\Delta G > 0$,结冰非自发 (冰会融化)。
- 如果 $\Delta G = 0$,冰水共存,处于平衡态 (此时 $T$ 为熔点)。 例如,在 $-10^\circ C (263 K)$ 时,$\Delta H = -6.01 \text{ kJ/mol}$,$\Delta S = -22 \text{ J/mol K} = -0.022 \text{ kJ/mol K}$。 $$ \Delta G = -6.01 \text{ kJ/mol} - (263 \text{ K})(-0.022 \text{ kJ/mol K}) $$ $$ \Delta G = -6.01 + 5.786 = -0.224 \text{ kJ/mol} $$ 因为 $\Delta G < 0$,所以在 $-10^\circ C$ 时水结冰是自发过程。
5. 描述过程:等温膨胀 (Describing a process)
(Slide 18)
- 考虑单原子理想气体在恒温 $T$、恒定粒子数 $N$ 条件下从 $p_i$ 膨胀到 $p_f$。计算吉布斯自由能的变化 $\Delta G$。
- 我们有 $dG = Vdp -SdT + \mu dN$。
- 由于 $T$ 和 $N$ 恒定,所以 $dT=0, dN=0$。 $$ dG = Vdp $$
- 积分得到 $\Delta G$: $$ \Delta G = \int_{p_i}^{p_f} V dp $$
- 对于理想气体,$pV=NkT \Rightarrow V = NkT/p$。 $$ \Delta G = \int_{p_i}^{p_f} \frac{NkT}{p} dp = NkT \int_{p_i}^{p_f} \frac{1}{p} dp $$ $$ \Delta G = NkT \ln\left(\frac{p_f}{p_i}\right) $$
平衡条件的策略 (Strategy for equilibrium conditions)
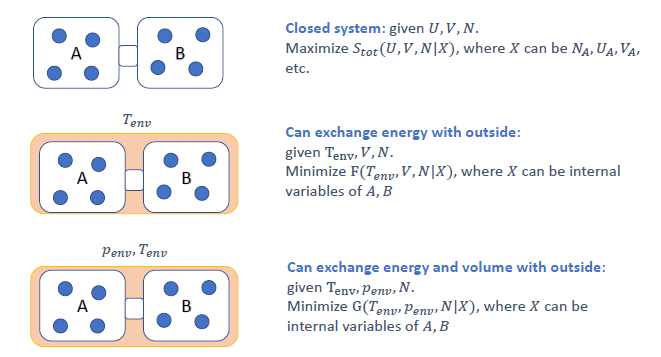
系统总是趋向于使其总熵 (系统+环境) 最大化的状态。自由能 $F$ 和 $G$ 是在特定约束条件下,仅用系统本身的性质来判断平衡和自发性的工具。
-
孤立系统 (Closed system) (恒定 $U, V, N$):
- 最大化总熵 $S_{tot}$ (Slide 23)。
- 这通常意味着系统内部达到均匀。
-
恒温恒容系统 (Constant T, V, N) (可以与外界交换能量):
-
最小化亥姆霍兹自由能 $F(T, V, N X)$,其中 $X$ 是内部变量 (例如,两相中的粒子数分配 $N_{gas}, N_{solid}$,如 Slide 21)。 - 策略 (Slide 21):
- 使用 $F = U - TS$。
- 找到可以变化的内部变量 $x$ (例如 $N_{gas}$,而 $N_{solid} = N_{total} - N_{gas}$)。
- 设置 $\frac{dF}{dx} = 0$,解出平衡条件。
-
-
恒温恒压系统 (Constant T, P, N) (可以与外界交换能量和体积):
-
最小化吉布斯自由能 $G(T, P, N X)$ (Slide 23)。 - 策略 (Slide 22):
- 使用 $G = U + pV - TS$。
- 找到可以变化的内部变量 $x$。
- 设置 $\frac{dG}{dx} = 0$,解出平衡条件。
-
总结各种热力学势 (Slide 20, 28)
| 量 (Quantity) | 定义 (Definition) | 微分 (Differential) | 控制变量 (Control variables) |
|---|---|---|---|
| 熵 (Entropy, S) | $S = k \ln \Omega$ | $dS = \frac{1}{T}dU + \frac{p}{T}dV - \frac{\mu}{T}dN$ | $U, V, N$ |
| 亥姆霍兹自由能 (F) | $F = U - TS$ | $dF = -SdT - pdV + \mu dN$ | $T, V, N$ |
| 吉布斯自由能 (G) | $G = U - TS + pV$ | $dG = -SdT + Vdp + \mu dN$ | $T, p, N$ |
以及最根本的内能微分形式: $$ dU = TdS - pdV + \mu dN $$
Lecture 12: Phases
核心概念:相、相图与化学势
1. 相与相图 (Phases and Phase Diagrams)
(Slides 2, 3, 10, 20, 21, 22)
-
什么是相 (Phase)? (Slide 3)
- 物质在其物理性质 (如密度、晶体结构) 在宏观上均匀一致的状态。常见的三相是固相 (Solid)、液相 (Liquid) 和气相 (Gas)。
-
微观差异:
- 固相: 原子/分子有固定的相对位置,排列有序。熵 (Entropy) $S$ 较低。
- 液相: 原子/分子间关系较松散,仍有一定关联,但可以流动。熵比固相高。
- 气相: 原子/分子间几乎没有关联,运动自由。熵通常远大于液相和固相。
-
相图 (Phase Diagram) (Slides 2, 9, 10, 20, 21, 22)
- 通常是压力-温度 ($p-T$) 图,显示在不同 $p, T$ 条件下物质的稳定相态。
- 相界线 (Phase boundaries): 图中的线表示两种相可以共存 (处于平衡) 的 $p, T$ 条件。例如,液-气界线是沸点随压力的变化曲线。
- 三相点 (Triple point): 三条相界线交汇于一点,表示固、液、气三相可以同时平衡共存的唯一 $p, T$ 条件。 (Slide 9, 10, 22)
- 临界点 (Critical point): 液-气相界线的终点。超过此点,液相和气相无法区分,物质处于超临界流体 (Supercritical fluid) 状态。(Slide 22 for CO2)
- 普遍规律: 在 $p-T$ 平面的大部分区域,只有一种相是稳定的 (Slide 10)。
2. 相平衡的热力学判据:吉布斯自由能与化学势
(Slides 4, 5, 6, 7, 8, 9)
-
恒温恒压下的平衡 (Slide 4):
- 在恒定的温度 $T$ 和压力 $p$ 条件下,系统达到平衡时,其吉布斯自由能 (Gibbs Free Energy) $G$ 最小。
- 对于一个可以存在于多个相态 (如气相、液相、固相) 的纯物质,其总吉布斯自由能为: $$ G = \mu_{gas}N_{gas} + \mu_{liquid}N_{liquid} + \mu_{solid}N_{solid} $$ 其中 $\mu_i$ 是第 $i$ 相的化学势 (Chemical Potential),$N_i$ 是第 $i$ 相的粒子数。
- 核心判据: 为了使 $G$ 最小,粒子会自发地从化学势高的相转移到化学势低的相。因此,在平衡时,所有共存相的化学势必须相等。如果只有一个相稳定,那么该相的化学势是所有可能相中最低的。
-
化学势 $\mu$ 对 $T$ 和 $p$ 的依赖关系 (Slide 6):
- 我们知道对于单组分系统,$G = N\mu$。其微分形式为 $dG = Vdp - SdT$ (当 $N$ 固定时)。
- 因此,$N d\mu = Vdp - SdT$ (这里假设 $N$ 固定,我们考察的是单位粒子或单位摩尔量的化学势如何随 $T,P$ 变化)。
- 所以,化学势的微分形式为: $$ d\mu = \left(\frac{V}{N}\right)dp - \left(\frac{S}{N}\right)dT = v dp - s dT $$ 其中 $v = V/N$ 是单位粒子体积 (或摩尔体积),$s = S/N$ 是单位粒子熵 (或摩尔熵)。
- 这意味着:
- $\left(\frac{\partial \mu}{\partial p}\right)_T = v$: 在恒温下,化学势随压力的变化率等于单位粒子体积。因为 $v > 0$,所以 $\mu$ 随 $p$ 的增加而增加。
- $\left(\frac{\partial \mu}{\partial T}\right)_p = -s$: 在恒压下,化学势随温度的变化率等于负的单位粒子熵。因为 $s > 0$,所以 $\mu$ 随 $T$ 的增加而减小。
-
理解相界线 (Slides 7, 8, 9):
-
$\mu$ vs. $p$ 曲线 (恒T) (Slide 7):
- 曲线的斜率是 $v$。密度更高的相 ($v$ 更小) 的 $\mu-p$ 曲线斜率更小。
- 当两条 $\mu-p$ 曲线相交时,意味着两相在该压力下化学势相等,可以共存。
-
$\mu$ vs. $T$ 曲线 (恒P) (Slide 8):
- 曲线的斜率是 $-s$。熵越大的相,其 $\mu-T$ 曲线的斜率越负 (即下降得越陡峭)。
- 通常 $s_{gas} > s_{liquid} > s_{solid}$,所以气相的 $\mu-T$ 曲线最陡峭,固相最平缓。
- Slide 8 的问题:哪个曲线可以代表一个普适物理系统?所有曲线的斜率都应该是负的。且因为 $s$ 随 $T$ 增加 (因为 $c_p > 0$, $(\partial s/\partial T)_p = c_p/T$),所以 $-s$ 应该变得更负,即曲线是向下凹的 (concave down)。图中的曲线 (a), (b), (c) 都满足向下凹且斜率为负。因此 (d) All of the above 可能是预期的答案,因为它们都可能代表某一相在一定温度范围内的行为。
-
从 $\mu-T$ 曲线构建 $p-T$ 相图 (Slide 9):
- 对于固定的压力 $p_1, p_2, p_3$,画出各相的 $\mu-T$ 曲线。
- 在任一温度 $T$,化学势最低的相是稳定相。
- 曲线的交点对应相变温度。例如,在 $p_1$ 下,$T_1$ 是升华点 (固-气转变)。
- 改变压力会移动这些 $\mu-T$ 曲线 (尤其是气相的,因为它对压力敏感)。例如,增加压力会显著增加 $\mu_{gas}$ (理想气体 $\mu_{gas} = kT \ln(p/p_Q)$),使其曲线向上移动。这会导致相变温度的变化。
- 三条曲线在一点相交的 $(T, p)$ 组合即为三相点 (例如图中的 $T_2, p_2$)。
-
$\mu$ vs. $p$ 曲线 (恒T) (Slide 7):


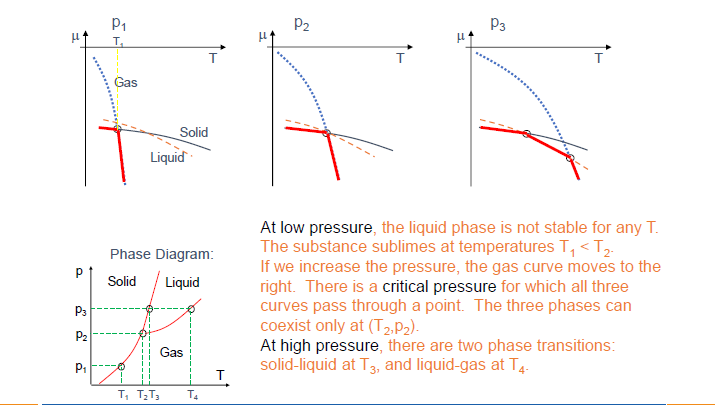
-
理想气体的化学势回顾 (Slide 5):
$$
\mu_g = kT \ln\left(\frac{n}{n_Q}\right) = kT \ln\left(\frac{p}{p_Q}\right)
$$
其中量子浓度 $n_Q \propto T^{3/2}$,所以量子压力 $p_Q = n_Q kT \propto T^{5/2}$。
- 通常情况下 ($n \ll n_Q$ 或 $p \ll p_Q$),$\mu_g$ 是负的。
- $\mu_g$ 随压力 $p$ 对数增加,随温度 $T$ 近似线性增加 (实际上由于 $p_Q$ 对 $T$ 的依赖,关系更复杂,导致 $\mu-T$ 曲线向下弯曲,如图中所示,恒压时 $\mu$ 随 $T$ 增加而减少)。
- Slide 5 的图示:恒压下,温度升高,$\mu$ 减小。恒温下,压力升高,$\mu$ 增大。
相变潜热 (Latent Heat) 与焓 (Enthalpy)
-
潜热 (Latent Heat, $Q_L$ 或 $L$) :
- 在相变过程中,物质在温度不变的情况下吸收或放出的热量。
- 例如,在恒压下加热液体,当达到沸点时,继续加热,温度保持不变,直到所有液体都变成气体。这期间吸收的热量就是汽化潜热。
- 从热力学第一定律 $Q = \Delta U + p\Delta V$ (恒压过程)。这部分能量用于增加内能 (克服分子间作用力) 和对外做膨胀功。
-
焓 (Enthalpy, $H$) :
- 定义: $H = U + pV$。
- 在恒压过程中,系统吸收的热量等于其焓变: $Q_p = \Delta H$。(热力学第一定律)
- 因此,相变潜热等于相变过程中的焓变: $$ Q_L = \Delta H_{transition} $$ 例如,汽化热 $\Delta H_{lg} = H_{gas} - H_{liquid}$。
- 相变过程也是熵变过程,在可逆相变 (温度恒定) 时: $$ \Delta S = \frac{Q_L}{T} = \frac{\Delta H}{T} $$
-
熵变的计算示例 (Slide 13): 水在 1 atm 结冰时的摩尔熵变。
- $Q_L$ (熔化热,从冰到水) $= 333 \text{ J/g}$。结冰时是放出热量,所以 $\Delta H_{freezing} = -333 \text{ J/g}$。
- $T = 0^\circ C = 273 \text{ K}$。
- 摩尔质量 $M = 18 \text{ g/mol}$。
- $\Delta S_{freezing, molar} = \frac{-333 \text{ J/g} \times 18 \text{ g/mol}}{273 \text{ K}} \approx -22.0 \text{ J/(mol K)}$。
- 负号表示熵减少,符合从无序度较高的液相到有序度较高的固相的转变。
- 每个水分子的玻尔兹曼熵 $\sigma = S/N_A k_B$ 变化约为 $-2.73$。这意味着液态时每个分子对应的微观状态数大约是固态时的 $e^{2.73} \approx 15$ 倍。
相变过程中相关量变化
Lecture12 Homework2 平衡温度、恒压状态下
- $\Delta H=\Delta Q$
- $\Delta S =\frac{\Delta H}{T}$
- $\Delta G=0$ (相变点两种相处于平衡状态)
- 体积变化 $\Delta V= \frac{\Delta NkT}{p}$
-
内能变化 $\Delta U = \Delta H-p\Delta V$
- Lecture12 Homework3
影响相变温度的因素
1. 压力对沸点/凝固点的影响 (Clausius-Clapeyron Equation)
(Slide 14)

-
沸点与压力:
- 降低压力会降低气相的化学势 $\mu_g$。
- 在 $\mu-T$ 图上,$\mu_g(T)$ 曲线下移 (或其与 $\mu_l(T)$ 的交点左移)。
- 这导致液-气平衡 (沸腾) 发生在更低的温度。所以高海拔地区水的沸点较低。
- Slide 14 比较了 1 atm 和 0.8 atm 下水的沸点,结论是 $T_{boil}(0.8 \text{ atm}) < T_{boil}(1 \text{ atm})$。
-
克劳修斯-克拉佩龙方程 (Clausius-Clapeyron Equation) (Slide 14 提及了一个简化形式):
- 定量描述相界线上压力随温度的变化率: $$ \frac{dp}{dT} = \frac{L}{T\Delta v} = \frac{\Delta s}{\Delta v} $$ 其中 $L$ 是相变潜热 (每摩尔或每单位质量),$\Delta v$ 是相变过程中的单位粒子体积 (或摩尔体积/比容) 变化,$T$ 是相变温度。
- Slide 14 给出的 $p = p_0 e^{-L/kT}$ 是一个积分后的近似形式,通常用于固/液-气相变,假设潜热 $L$ 是常数,且气相体积远大于固/液相体积,并把气体视为理想气体。这里的 $L$ 是单个分子的潜热。
2. 溶质对凝固点/沸点的影响 (依数性)
(Slides 15, 16, 17)
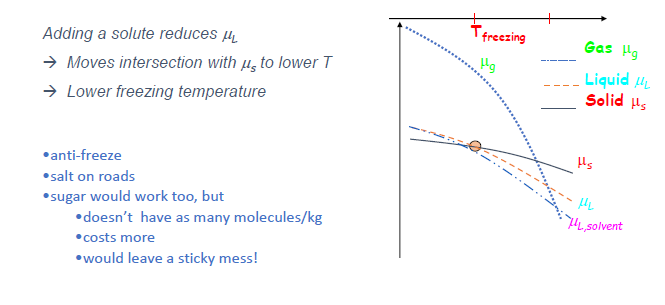
-
基本原理 (Slide 15):
- 向纯溶剂中溶解非易失性溶质 (solute) 会降低溶剂的化学势 $\mu_L$ (即 $\mu_{solvent, solution} < \mu_{pure solvent}$)。
- 这是因为溶解过程是自发的,意味着混合态的吉布斯自由能更低。
- 如果溶质不进入固相或气相,那么 $\mu_S$ 和 $\mu_G$ (纯溶剂的) 不受影响。
-
凝固点降低 (Freezing point depression) (Slide 16):
- 由于溶液中溶剂的 $\mu_L$ 降低了,其 $\mu_L(T)$ 曲线整体下移。
- 这使得 $\mu_L(T)$ 与固相纯溶剂的 $\mu_S(T)$ 曲线的交点向更低的温度移动。
- 因此,溶液的凝固点低于纯溶剂的凝固点。例子:防冻剂、路上撒盐化冰。
-
沸点升高 (Boiling point elevation):
- 类似地,$\mu_L(T)$ 曲线下移,导致其与气相纯溶剂的 $\mu_G(T)$ 曲线的交点向更高的温度移动。
- 因此,溶液的沸点高于纯溶剂的沸点 (前提是溶质非易失性)。
-
溶质对溶剂化学势影响的推导 (Slide 17):
- 假设溶剂 ($L$) 和溶质 ($S$) 形成理想溶液,混合过程的熵变主要是构型熵 (entropy of mixing)。
- 混合熵 (对于溶剂和溶质的总系统): $S_{mix} = k \ln \frac{(N_L + N_S)!}{N_L! N_S!}$ (这里 $N_L, N_S$ 是溶剂和溶质的粒子数)。
- 溶剂化学势的变化 $\Delta \mu_L = \mu_{L,solution} - \mu_{L,pure}$。可以通过吉布斯-亥姆霍兹关系或者直接考虑混合自由能得到。
- Slide 17 用了一个近似的方法来估计 $\Delta \mu_L$ 由于混合熵带来的变化: $\Delta \mu_{L(mix)} = -T \left(\frac{\partial S_{mix}}{\partial N_L}\right){N_S}$ (更准确的应该是考虑吉布斯自由能 $G{mix} = N_L \mu_{L,pure} + N_S \mu_{S,pure} - T S_{mix}$,然后 $\mu_{L,solution} = (\partial G_{mix}/\partial N_L)$)。
- 对于稀溶液 ($N_S \ll N_L$),利用斯特林近似,可以得到: $$ \Delta \mu_L \approx -kT \frac{N_S}{N_L} = -kT x_S $$ 其中 $x_S = N_S/(N_L+N_S) \approx N_S/N_L$ 是溶质的摩尔分数。
- 因为 $x_S > 0$,所以 $\Delta \mu_L < 0$,即溶剂在溶液中的化学势降低了。
亚稳态:过冷与过热
(Slide 18)
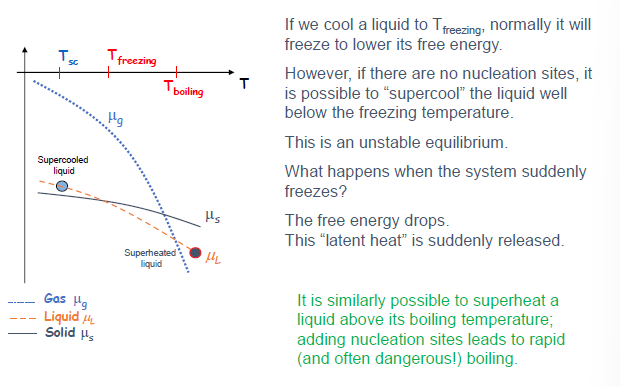
- 过冷 (Supercooling): 将液体冷却到其正常凝固点以下而不发生凝固的现象。
- 过热 (Superheating): 将液体加热到其正常沸点以上而不发生沸腾的现象。
- 原因: 相变通常需要形核点 (nucleation sites) 来启动。如果没有形核点,系统可以暂时处于亚稳态 (unstable equilibrium)。
- 在 $\mu-T$ 图上,亚稳态的相的化学势并不是最低的,但系统被困在这个状态。
- 一旦发生相变 (例如通过引入形核点或扰动),系统会迅速转变为更稳定的相 (化学势更低的相),并释放潜热,导致温度可能回升 (过冷液体凝固) 或剧烈沸腾 (过热液体沸腾)。
相变的一般规则
(Slides 19, 21)
-
压力效应: 增加压力有利于体积更小 (即密度更高) 的相。
- 因为 $(\partial \mu / \partial p)_T = v$。压力增大时, $v$ 小的相的 $\mu$ 增加得慢,更容易成为低 $\mu$ 的稳定相。
- 例如,对于大多数物质 (如氨,Slide 21),固相密度 > 液相密度 > 气相密度。所以高压有利于固相。水是例外,冰的密度小于水。
-
温度效应: 增加温度有利于熵更大 (即无序度更高) 的相。
- 因为 $(\partial \mu / \partial T)_p = -s$。温度升高时, $s$ 大的相的 $\mu$ 下降得快,更容易成为低 $\mu$ 的稳定相。
- 通常 $s_{gas} > s_{liquid} > s_{solid}$。所以高温有利于气相。
二元相图 (Binary phase diagrams)
(Slide 23)
- 这是对双组分系统 (如合金) 相平衡的介绍。
- 相图变得更复杂,除了 $T, p$ 外,还有一个变量是成分浓度。
- 图中可能出现共晶点 (Eutectic point)、固溶体 (Solid solution, 如 $\alpha, \beta$) 等。
- 这部分内容通常在材料科学或更高等的热力学课程中详细讨论,这里只是一个初步的展示,表明相图可以描述更复杂的系统。